Mit künstlicher Intelligenz zum chemischen Fingerabdruck
26. September 2017
Die Forscher haben mittels künstlicher Intelligenz einen Weg gefunden, chemische Simulationen massiv zu beschleunigen (Copyright: Michael Gastegger).
Neuronale Netze rechnen chemische Simulationen in Rekordzeit
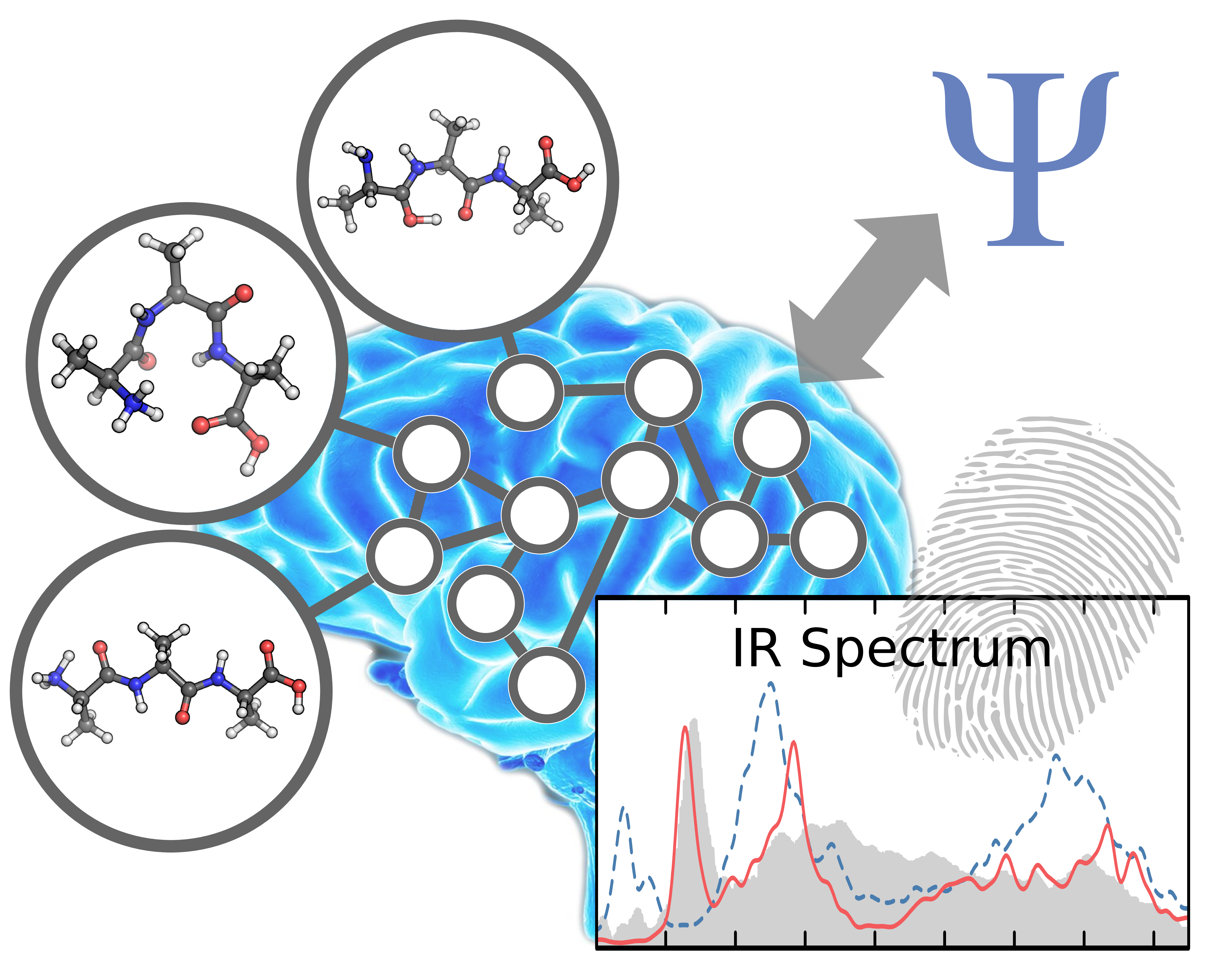
Forschern an den Universitäten Wien und Göttingen ist es gelungen, eine auf künstlicher Intelligenz aufbauende Methode zur Vorhersage von molekularen Infrarotspektren zu entwickeln. Diese chemischen "Fingerabdrücke" konnten von gängigen Vorhersagetechniken bislang nur für kleine Moleküle in hoher Qualität simuliert werden. Mit Hilfe der neuen Technik, die auf neuronalen Netzwerken ähnlich dem menschlichen Gehirn basiert und damit lernfähig ist, konnte das Team um Philipp Marquetand von der Fakultät für Chemie der Universität Wien Simulationen durchführen, die zuvor nicht möglich waren. Das Potenzial dieser neuen Strategie wurde nun in der aktuellen Ausgabe der Fachzeitschrift "Chemical Science" publiziert.
Drastische Fortschritte in der Forschung zu künstlicher Intelligenz haben im letzten Jahrzehnt zu einer großen Reihe von faszinierenden Entwicklungen in diesem Bereich geführt. Selbstständig fahrende Autos, aber auch alltägliche Anwendungen wie Suchmaschinen und Spam-Filter veranschaulichen die vielseitige Einsetzbarkeit von Methoden aus dem Gebiet der künstlichen Intelligenz.
Infrarotspektroskopie ist eine der wertvollsten experimentellen Methoden, um Einblick in die Welt der Moleküle zu erhalten. Infrarotspektren sind chemische Fingerabdrücke, welche Aufschluss über die Zusammensetzung und Eigenschaften von Substanzen und Materialien geben. In vielen Fällen sind diese Spektren sehr komplex – eine detaillierte Analyse macht computergestützte Simulationen unumgänglich. Während quantenchemische Rechnungen im Prinzip eine äußerst exakte Vorhersage von Infrarotspektren ermöglichen, wird ihre Anwendbarkeit in der Praxis durch den mit ihnen verbundenen hohen Rechenaufwand erschwert. Aus diesem Grund können verlässliche Infrarotspektren nur für relativ kleine chemische Systeme berechnet werden.
Eine internationale Gruppe von Forschern unter der Leitung von Philipp Marquetand von der Fakultät für Chemie der Universität Wien hat nun einen Weg gefunden, diese Simulationen mittels künstlicher Intelligenz zu beschleunigen. Zu diesem Zweck werden sogenannte künstliche neuronale Netzwerke verwendet, mathematische Modelle des menschlichen Gehirns. Diese sind in der Lage, die komplexen quantenmechanischen Beziehungen, die zur Modellierung von Infrarotspektren nötig sind, anhand einiger weniger Beispiele zu lernen. Auf diese Art und Weise können die Wissenschafter Simulationen innerhalb weniger Minuten durchführen, die sonst selbst mit modernen Supercomputern Jahrtausende in Anspruch nehmen würden – ohne dabei an Verlässlichkeit einzubüßen. "Wir können nun endlich chemische Problemstellungen simulieren, die mit den bis dato verwendeten Simulationstechniken nicht zu bewältigen waren", sagt der Erstautor der Studie, Michael Gastegger.
Aufgrund der Ergebnisse dieser Studie sind die Forscher zuversichtlich, dass ihre Methode zur Spektrenvorhersage in Zukunft weitreichenden Einsatz in der Analyse von experimentellen Infrarotspektren finden wird.
Publikation in "Chemical Science"
Machine learning molecular dynamics for the simulation of infrared spectra
Michael Gastegger, Jörg Behler, Philipp Marquetand
Chemical Science, 2017.
DOI:10.1039/C7SC02267K
Wissenschaftlicher Kontakt
Priv.-Doz. Dr. Philipp Marquetand
Institut für Theoretische ChemieUniversität Wien
1090 - Wien, Währinger Straße 17
+43-1-4277-527 64
philipp.marquetand@univie.ac.at
Rückfragehinweis
Stephan Brodicky
Pressebüro der Universität WienUniversität Wien
1010 - Wien, Universitätsring 1
+43-1-4277-175 41
+43-664-60277-175 41
stephan.brodicky@univie.ac.at
Downloads:
press_releaseIMG_01.png
{kind=link}
Dateigröße: 2,43 MB