Ein "molekularer" Kompass zeigt den Weg zur Reduzierung von Tierversuchen
20. August 2024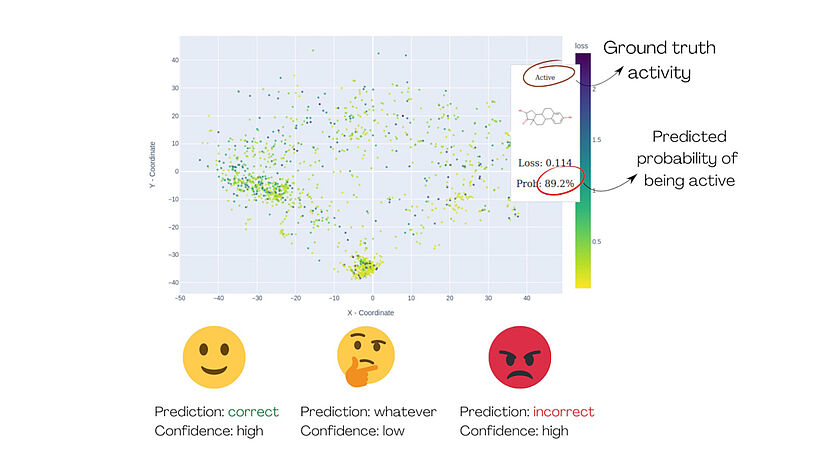
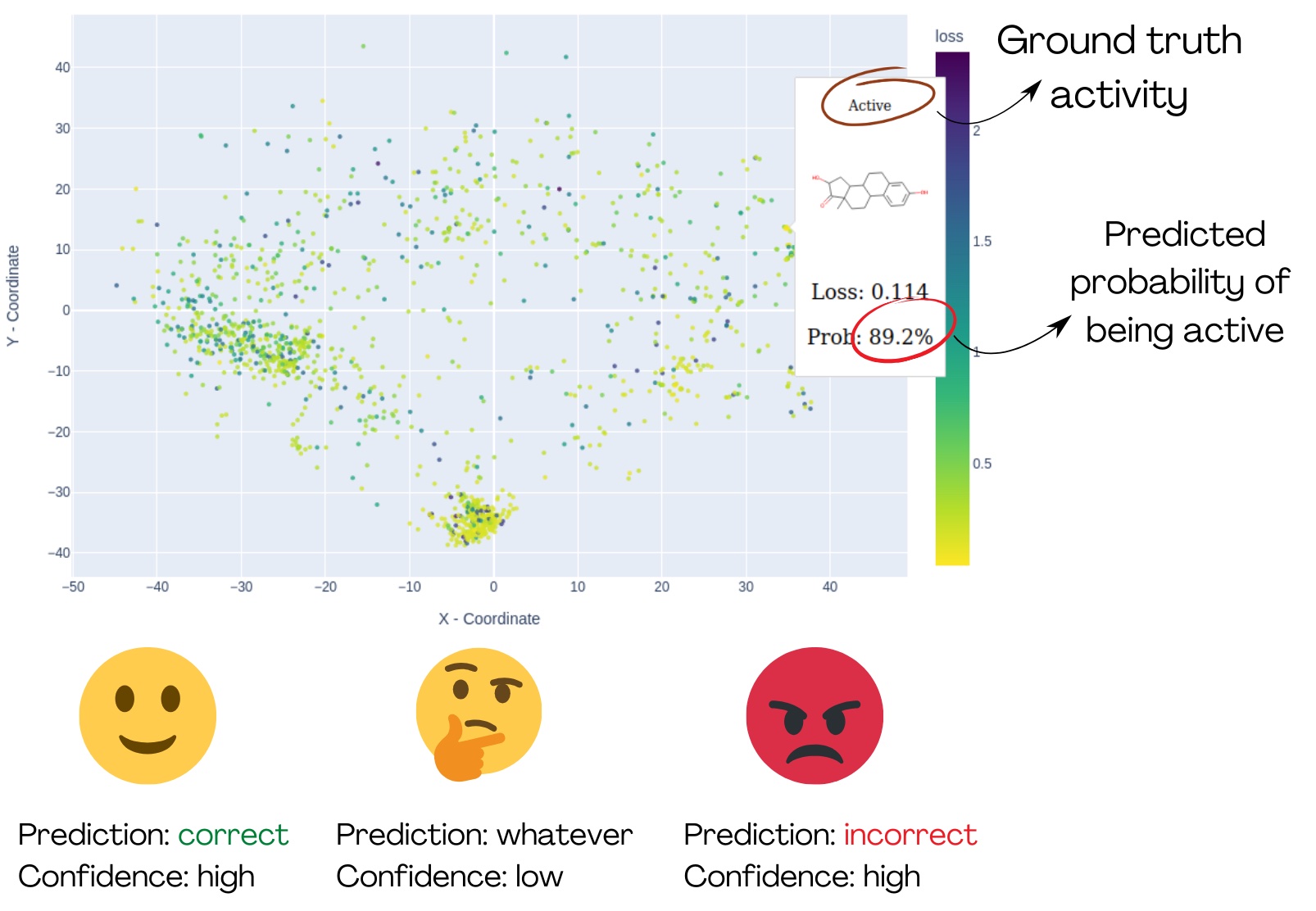
Abb. 1: Die Demonstration von MolCompass veranschaulicht, wie ein Computertoxikolog*innen die betreffenden Bereiche des chemischen Raums identifizieren können. Mit Hilfe der Software können die Toxikolog*innen Regionen ausfindig machen, in denen das untersuchte Modell die Aktivität mit hoher Sicherheit falsch vorhersagt. C: Sergey Sosnin
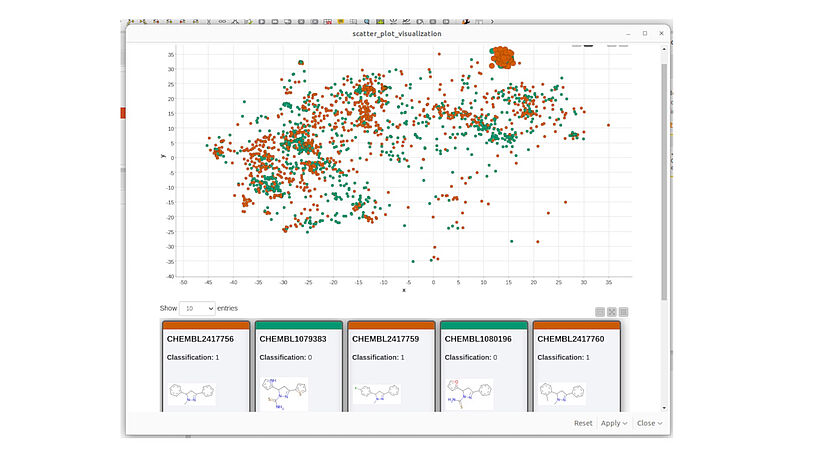
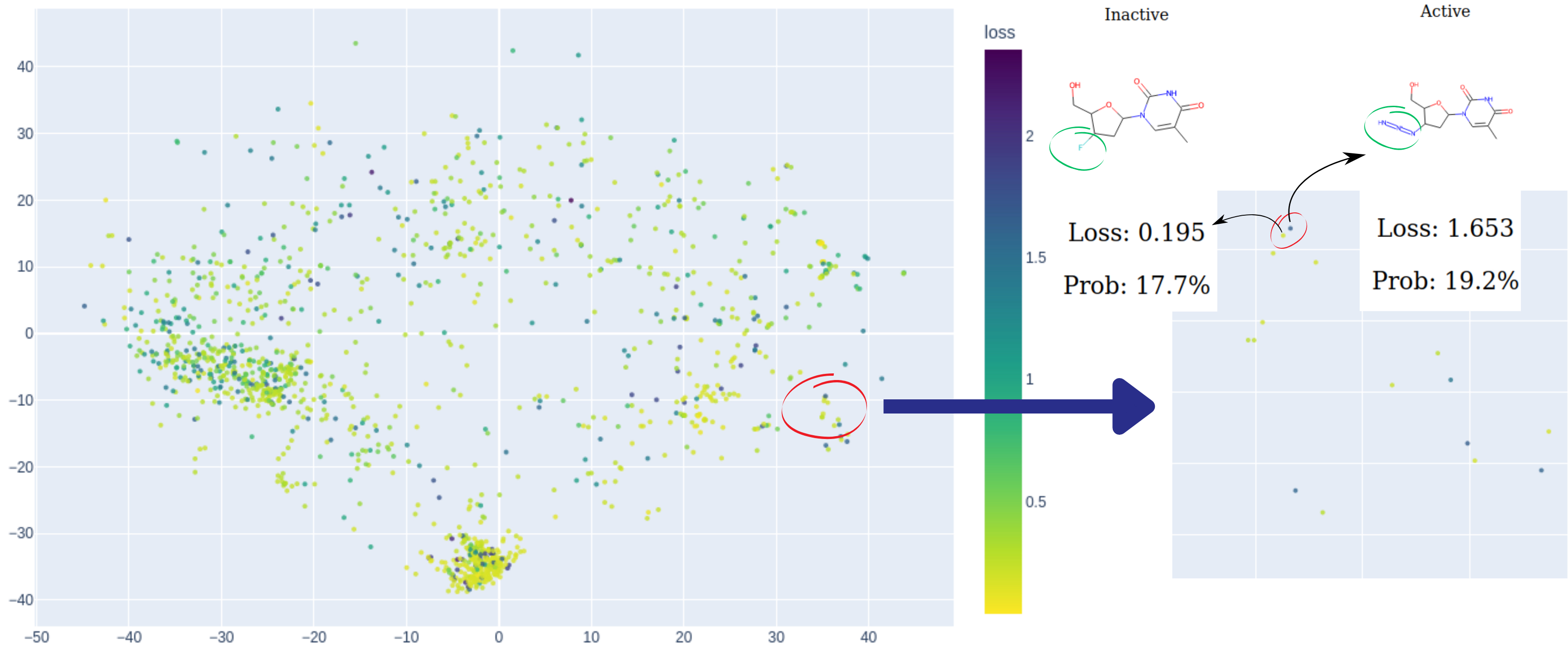
Abb. 2: Eine Illustration der Suche nach Modellklippen. Auf der linken Seite der chemischen Karte liegen zwei Punkte nahe beieinander, weisen jedoch unterschiedliche Farben auf. Eine weitere Untersuchung dieser merkwürdigen Beobachtung zeigt, dass diese beiden Verbindungen zwar ein hohes Maß an struktureller Ähnlichkeit aufweisen, aber gegensätzliche Aktivitäten entfalten, was eine Herausforderung darstellt, die das Modell nicht effektiv lösen kann. Die visualisierten Daten sind: Östrogene Bindemittel-Datensatz. C: Sergey Sosnin
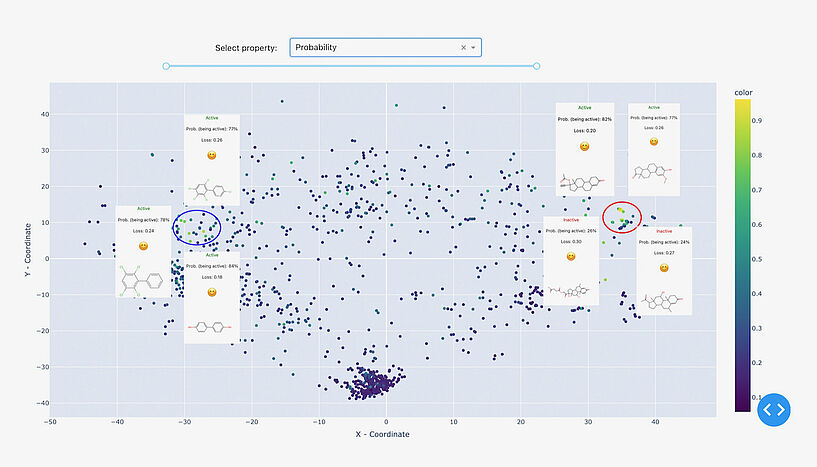
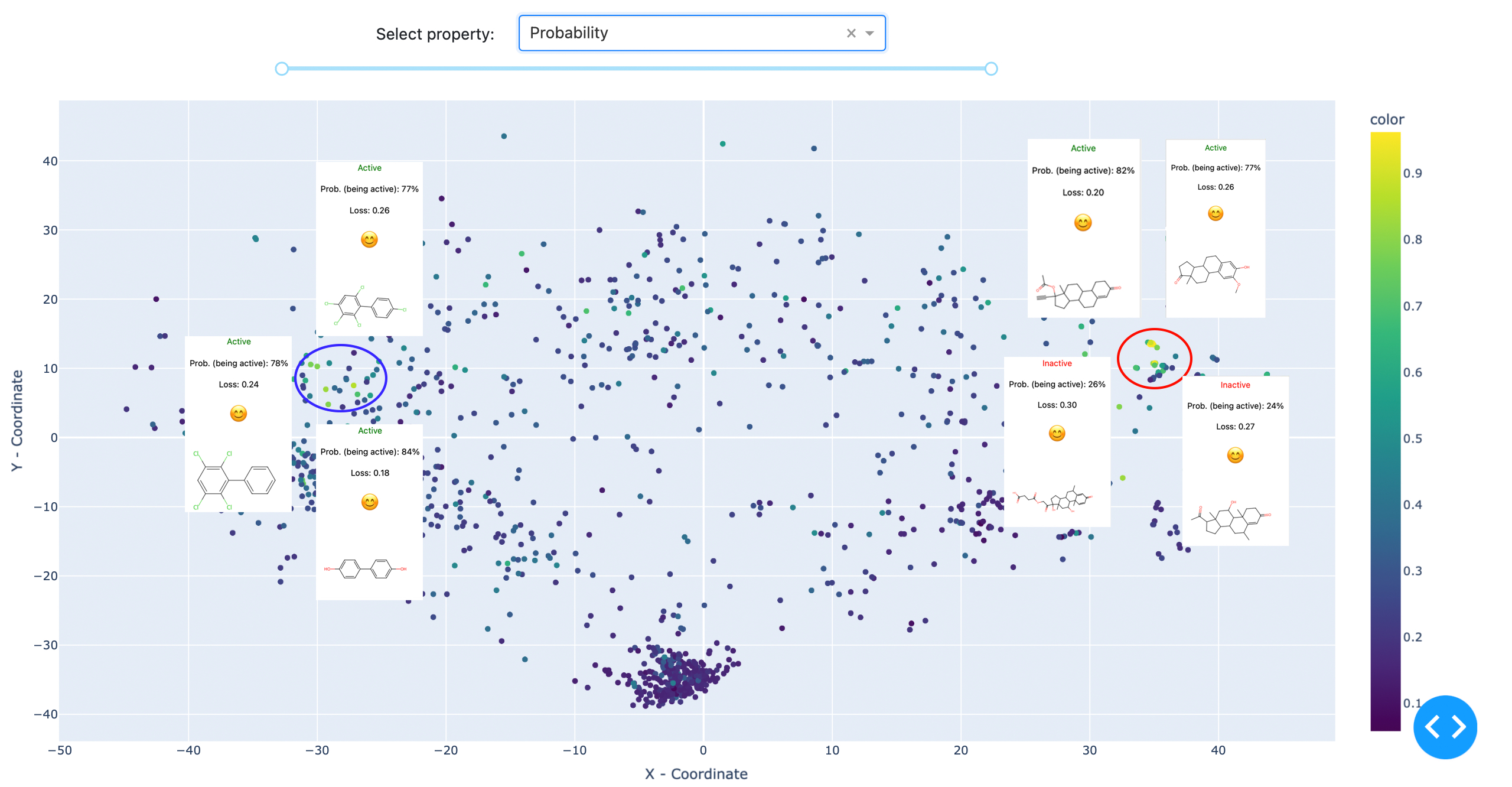
Abb. 3: Zwei Cluster wurden vom Referenzmodell mit hoher Sicherheit zugeordnet. Der dichtere Cluster auf der linken Seite steht für Steroidderivate, während der rechte, weniger definierte Cluster polychlorierte Biphenyle und Polyphenole umfasst. Die visualisierten Daten sind: Östrogene Bindemittel-Datensatz. C: Sergey Sosnin
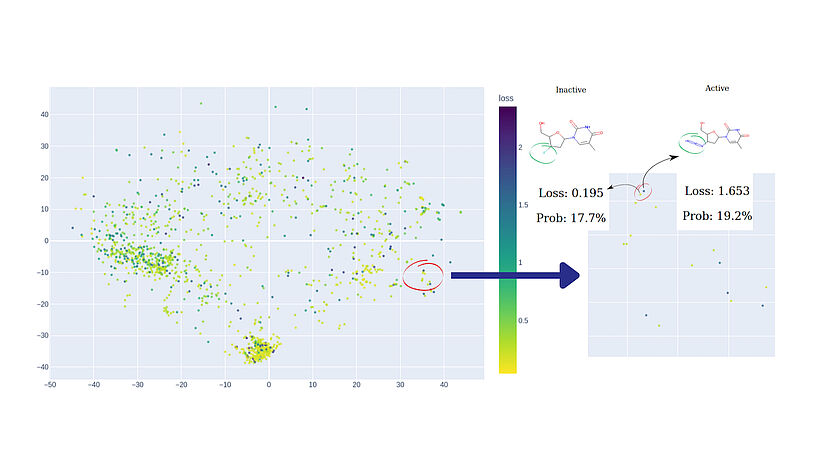
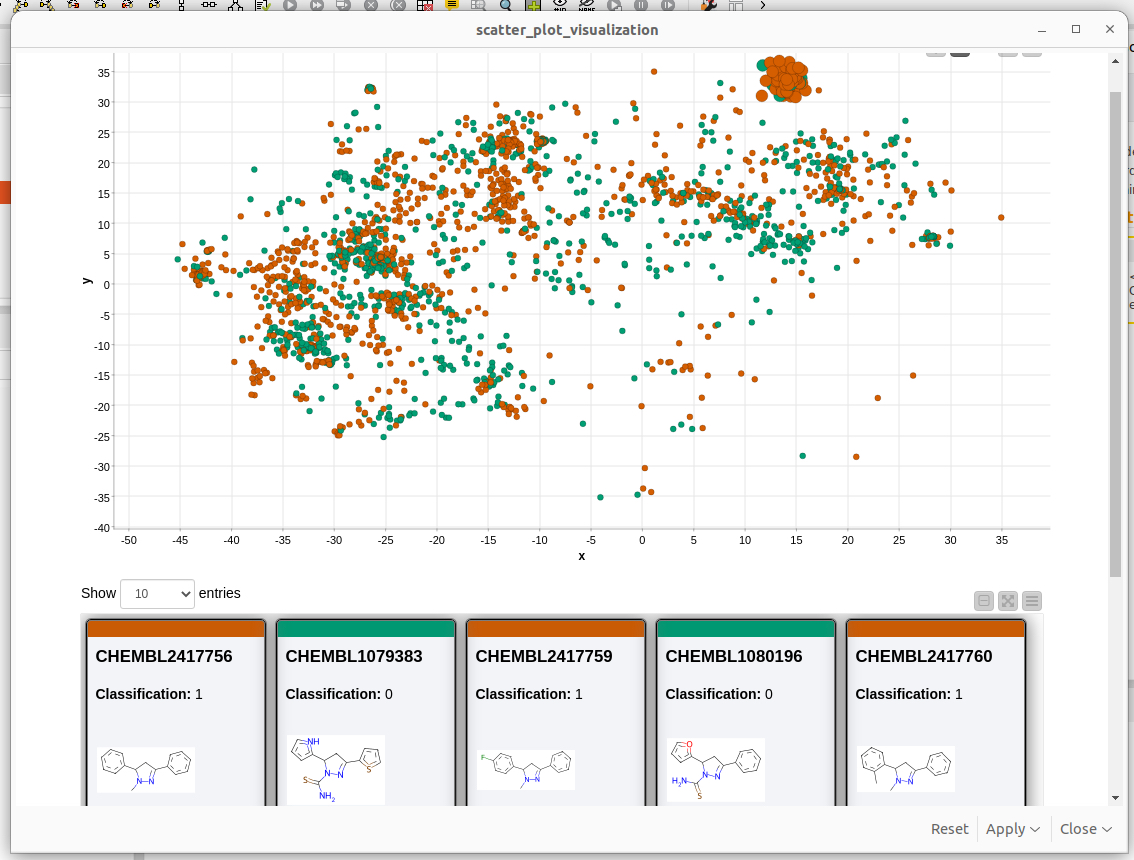
Abb. 4: Ein Screenshot der KNIME-Visualisierung des chemischen Raums mit dem MolCompass KNIME-Knoten. C: Sergey Sosnin
Wissenschafter*innen entwickeln intelligentes Software-Tool zur Bewertung chemischer Risiken
In den vergangenen Jahren sind Machine Learning Methoden zur Risikobewertung chemischer Verbindungen immer wichtiger geworden. Sie sind aber auch eine "Black Box" aufgrund fehlender Nachvollziehbarkeit und Transparenz, was zu Skepsis unter Fachleuten und Zulassungsbehörden führt. Um das Vertrauen in diese Modelle zu erhöhen, haben Forscher*innen der Universität Wien jene Bereiche identifiziert, in denen diese Modelle Schwächen aufweisen. Zu diesem Zweck entwickelten sie ein innovatives Software-Tool ("MolCompass"). Die Ergebnisse dieses Forschungsansatzes wurden gerade im renommierten Journal of Cheminformatics veröffentlicht.
Über viele Jahrzehnte wurden neue Arzneimittel und Agrarchemikalien hauptsächlich an Tieren getestet. Diese Tests sind teuer, werfen ethische Bedenken auf und versagen oft bei der genauen Vorhersage von Nebenwirkungen am Menschen. Im Rahmen des von der Europäischen Union unterstützten Projektes RISK-HUNT3R wird – unter Mitarbeit von Wissenschafter*innen der Universität Wien – an der Entwicklung der nächsten Generation von Methoden zur tierversuchsfreien Risikobewertung neuer Substanzen geforscht. Rechnergestützte Methoden ermöglichen es mittlerweile, die toxikologischen und ökologischen Risiken neuer Chemikalien vollständig per Computer zu bewerten, ohne dass die chemischen Verbindungen synthetisiert und getestet werden müssen. Aber eine Frage bleibt: Wie vertrauenswürdig sind diese Computermodelle?
Es geht um zuverlässige Vorhersagen
Um dieses Problem näher zu untersuchen konzentrierte sich Sergey Sosnin, Senior Scientist in der Forschungsgruppe für Pharmakoinformatik an der Universität Wien, auf die binäre Klassifikation. Hierbei liefert ein maschinelles Lernmodell eine Wahrscheinlichkeit von 0 % bis 100 %, die angibt, ob eine chemische Verbindung aktiv ist oder nicht (z. B. toxisch oder nicht toxisch, bioakkumulierbar oder nicht bioakkumulierbar, ein Binder oder Nicht-Binder an ein spezifisches menschliches Protein). Diese Wahrscheinlichkeit spiegelt das Vertrauen des Modells in seine Vorhersage wider. Idealerweise sollte das Modell nur bei korrekten Vorhersagen Werte nahe 0% (sicher inaktiv) oder 100% (Sicher aktiv) geben. Wenn das Modell unsicher ist und eine Vertrauensbewertung von z.B. 51 % abgibt, sollten diese Vorhersagen verworfen und alternative Methoden zur Risikobewertung herangezogen werden. Ein Problem entsteht jedoch dann, wenn das Modell falsche Vorhersagen mit hohen Wahrscheinlichkeiten liefert.
"Dies ist das wahre Albtraumszenario für Toxikolog*innen", sagt Sergey Sosnin. "Wenn ein Modell vorhersagt, dass eine Verbindung mit 99 % Sicherheit nicht toxisch ist, die Verbindung aber tatsächlich toxisch ist, gibt es keine Möglichkeit zu wissen, dass etwas falsch gelaufen ist." Die einzige Lösung besteht darin, jene Bereiche des chemischen Raums – also mögliche Klassen organischer Verbindungen – im Voraus zu identifizieren, in denen das Modell "blinde Flecken" hat, und diese zu vermeiden. Dazu müssen Forscher*innen, die das Modell bewerten, die vorhergesagten Ergebnisse für Tausende von chemischen Verbindungen einzeln überprüfen – eine mühsame und fehleranfällige Aufgabe.
Überwindung dieses bedeutenden Hindernisses
"Um diese Forschenden zu unterstützen", fährt Sosnin fort, "entwickelten wir interaktive grafische Werkzeuge, die chemische Verbindungen auf eine 2D-Ebene projizieren, ähnlich wie geografische Karten. Mit Farben heben wir die Verbindungen hervor, die mit hoher Sicherheit falsch vorhergesagt wurden, sodass Benutzer*innen sie als Cluster roter Punkte identifizieren können. Die Karte ist interaktiv und ermöglicht es den Benutzer*innen, den chemischen Raum zu untersuchen und besorgniserregende Bereiche zu erkunden."
Die Methodik wurde anhand eines Modells zur Bindung an den Östrogenrezeptor getestet. Nach der visuellen Analyse des chemischen Raums wurde klar, dass das Modell gut für z. B. Steroide und polychlorierte Biphenyle funktioniert, aber bei kleinen, nicht zyklischen Verbindungen völlig versagt und daher nicht für diese verwendet werden sollte.
Die in diesem Projekt entwickelte Software ist der wissenschaftlichen Community frei zugänglich auf GitHub verfügbar. Sergey Sosnin hofft, dass MolCompass Chemiker*innen und Toxikolog*innen zu einem besseren Verständnis der Einschränkungen von Computermodellen verhelfen wird. Diese Studie ist ein Schritt in Richtung einer Zukunft, in der Tierversuche nicht mehr notwendig sein werden und der einzige Arbeitsplatz für Toxikolog*innen ein Schreibtisch mit einem Rechner ist.
Originalpublikation:
S. Sosnin: MolCompass: multi-tool for the navigation in chemical space and visual validation of QSAR/ QSPR models. Journal of Cheminformatics.
DOI: 10.1186/s13321-024-00888-z
Abbildungen:
Abb. 1: Die Demonstration von MolCompass veranschaulicht, wie ein Computertoxikolog*innen die betreffenden Bereiche des chemischen Raums identifizieren können. Mit Hilfe der Software können die Toxikolog*innen Regionen ausfindig machen, in denen das untersuchte Modell die Aktivität mit hoher Sicherheit falsch vorhersagt. C: Sergey Sosnin
Abb. 2: Eine Illustration der Suche nach Modellklippen. Auf der linken Seite der chemischen Karte liegen zwei Punkte nahe beieinander, weisen jedoch unterschiedliche Farben auf. Eine weitere Untersuchung dieser merkwürdigen Beobachtung zeigt, dass diese beiden Verbindungen zwar ein hohes Maß an struktureller Ähnlichkeit aufweisen, aber gegensätzliche Aktivitäten entfalten, was eine Herausforderung darstellt, die das Modell nicht effektiv lösen kann. Die visualisierten Daten sind: Östrogene Bindemittel-Datensatz. C: Sergey Sosnin
Abb. 3: Zwei Cluster wurden vom Referenzmodell mit hoher Sicherheit zugeordnet. Der dichtere Cluster auf der linken Seite steht für Steroidderivate, während der rechte, weniger definierte Cluster polychlorierte Biphenyle und Polyphenole umfasst. Die visualisierten Daten sind: Östrogene Bindemittel-Datensatz. C: Sergey Sosnin
Abb. 4: Ein Screenshot der KNIME-Visualisierung des chemischen Raums mit dem MolCompass KNIME-Knoten. C: Sergey Sosnin
Wissenschaftlicher Kontakt
Sergey Sosnin, PhD
Department für Pharmazeutische WissenschaftenUniversität Wien
1090 - Wien, Josef-Holaubek-Platz
+43 1427755111
+49 1627541704
sergey.sosnin@univie.ac.at
Rückfragehinweis
Theresa Bittermann
Media Relations, Universität Wien1010 - Wien, Universitätsring 1
+43-1-4277-17541
theresa.bittermann@univie.ac.at
Downloads:
20240820_Sosnin_Abb4.jpg
{kind=link}
Dateigröße: 547,22 KB
20240820_Sosnin_Abb3.jpg
{kind=link}
Dateigröße: 1,81 MB
20240820_Sosnin_Abb2.jpg
{kind=link}
Dateigröße: 927,49 KB
20240820_Sosnin_Abb1.jpg
{kind=link}
Dateigröße: 242,38 KB