A new tool to decipher evolutionary biology
09. Mai 2017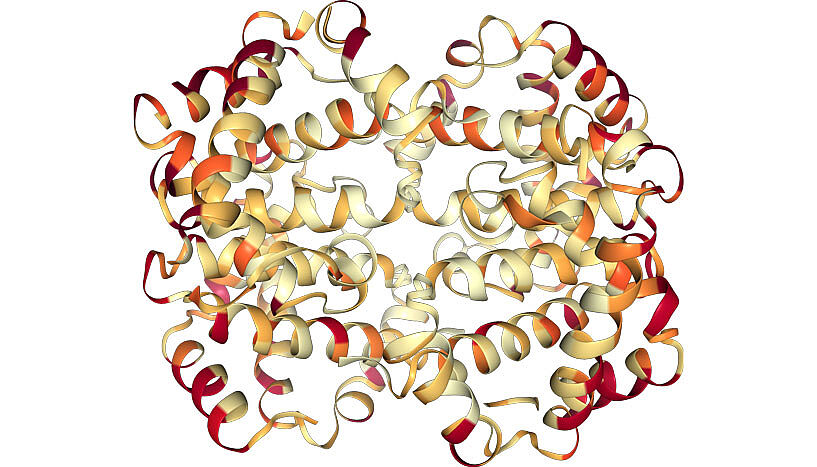
3D structure of human haemoglobin, showing residues with high (red) or low (light yellow) evolutionary rates as determined by ModelFinder (Copyright: Minh Quang Bui/University of Vienna)
A new bioinformatics tool to compare genome data has been developed by teams from the Max F. Perutz Laboratories, a joint venture of the University of Vienna and the Medical University of Vienna, together with researchers from Australia and Canada. The program called “ModelFinder” uses a fast algorithm and allows previously not attainable new insights into evolution. The results are published in the influential journal Nature Methods.
Understanding evolution is one of the cornerstones of biology – evolution is, in fact, the sole explanation for life’s diversity on our planet.
Based on the evolution of proteins, researchers may explain the emergence of new species and functions through genetic changes, how enzymes with novel functions might be engineered, or, for example, how humans are related to their closest relatives such as gorillas or bonobos.
One popular approach to the study of evolution is to compare genome data using bioinformatics (computer-aided) tools. Scientists using these approaches may compare specific proteins, which consist of combinations of 20 universal building blocks, called amino acids.
So far, the bioinformatics tools used to study the evolution of single proteins have assumed that the speed at which different regions of proteins evolve can be modeled with a statistical distribution whose shape is determined by a single variable.
“That assumption, however, does not reflect reality, and it might have led to a large proportion of biased phylogenetic results being published over the last two decades or so,” explains Minh Quang Bui, from the Center for Integrative Bioinformatics (CIBIV) and co-author of the study.
A new algorithm allows insights into protein evolution
Arndt von Haeseler, group leader at the Max F. Perutz Laboratories (MFPL) and Lars Jermiin from the Australian National University have now found a revolutionary way of implementing different rates of evolution into bioinformatics models.
It was well known among experts that the popular approach might not capture the complexities of protein evolution. However, the computational cost of using more realistic models was unacceptably high. “We have now developed a fast algorithm that gives us previously unavailable insights into protein evolution – the new tool is likely to have a huge impact on a wide variety of research areas, including on the evolution of pathogens and the dispersal of agricultural pests, ” adds Lars Jermiin.
The new program “ModelFinder” will allow more accurate scientific estimates of evolutionary processes. This enhanced understanding of evolution will help us come one step closer to unraveling the mysteries, which are responsible for the great diversity on our planet.
Service: Nature Methods
“ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates”. Subha Kalyaanamoorthy, Bui Quang Minh, Thomas KF Wong, Arndt von Haeseler, Lars S Jermiin. DOI:10.1038/NMETH.4285 (published online 8 May 2017)
Rückfragehinweis
Caterina Purini, MSc
Max F. Perutz Laboratories1030 - Wien, Dr.-Bohr-Gasse 9
+43-1-4277-240 14
+43-664-602 77-240 14
caterina.purini@mfpl.ac.at
Downloads:
hemoglobin_whole__c_Minh_Quang_Bui.png
{kind=link}
Dateigröße: 2,81 MB